
NOME DO EXAME
HBOC – Painel de Mama e Ovário – 25 genes – NGS com CNV
SÍNDROMES DE CÂNCER HEREDITÁRIO CONTEMPLADAS NESTE PAINEL
Painel de Predisposição Hereditária ao Câncer de Mama e Ovário; Câncer de mama; Painel de Câncer de Mama e Ovário; Síndrome de Cowden; HBOC; Câncer de Mama Hereditário; Câncer de Mama e Ovário Hereditário; Câncer Gástrico Difuso Hereditário; HNPCC; Síndrome de Li-Fraumeni; Síndrome de Lynch; Câncer de Ovário; Síndrome de Peutz-Jeghers; Síndrome do Tumor Hamartoma PTEN; Câncer endometrial;
DESCRIÇÃO
HBOC – A Síndrome Hereditária do Câncer de Mama e Ovário (HBOC) é uma condição genética que aumenta significativamente o risco de desenvolver câncer de mama, ovário e outros tipos de câncer. É mais comumente associada a mutações herdadas nos genes BRCA1 e BRCA2, embora outros genes contribuam com HBOC com diferentes graus de risco. Estes incluem os genes ATM, BARD1, BRIP1, CDH1, CHEK2, EPCAM, MLH1, MRE11 (MRE11A), MSH2, MSH6, NBN, NF1, PALB2, PMS1, PMS2, PTEN, RAD50, RAD51C, RAD51D, SMARCA4, STK11, TP53 e XRCC2. Esses genes desempenham papéis essenciais na manutenção da estabilidade genômica e na prevenção do câncer, e mutações neles podem causar várias síndromes hereditárias de câncer além de HBOC.
Sumário dos genes analisados
O gene ATM (Ataxia-Telangiectasia Mutada) está mapeado no cromossomo 11 (11q22) e codifica uma proteína quinase envolvida na detecção de danos ao DNA e ativação de vias de reparo. Esta proteína desempenha um papel fundamental na manutenção da estabilidade genômica. Mutações em ATM estão relacionadas ao câncer de mama, câncer de próstata e outros tipos de câncer, especialmente em indivíduos com síndrome de ataxia-telangiectasia. Mulheres com mutações ATM têm um risco elevado de câncer de mama.
O gene BARD1 (BRCA1-Associated RING Domain 1) está mapeado no cromossomo 2 (2q35) e codifica uma proteína que forma um complexo com BRCA1 e participa do reparo de DNA, regulação do ciclo celular e supressão de tumores. Mutações em BARD1 aumentam o risco de câncer de mama e podem contribuir para câncer de ovário e outros tipos de câncer, embora o risco seja menor do que o de BRCA1/BRCA2.
O gene BRCA1 (Breast Cancer 1) está mapeado no cromossomo 17 (17q21) e codifica uma proteína envolvida no reparo de DNA danificado, regulação do ciclo celular e manutenção da estabilidade genômica. Esta proteína desempenha um papel fundamental na recombinação homóloga, um processo crítico para consertar quebras de fita dupla de DNA. Mutações herdadas em BRCA1 aumentam significativamente o risco de desenvolver câncer de mama (particularmente câncer de mama de início precoce), câncer de ovário e outros tipos de câncer, incluindo câncer de trompa de Falópio e câncer peritoneal. Mulheres com mutação BRCA1 têm até 80% de risco vitalício de desenvolver câncer de mama e 40-60% de risco de desenvolver câncer de ovário.
O gene BRCA2 (Breast Cancer 2) está mapeado no cromossomo 13 (13q12) e codifica uma proteína envolvida no reparo de quebras de fita dupla de DNA por meio de recombinação homóloga. Esta proteína também ajuda a estabilizar a proteína RAD51, que desempenha um papel central no reparo do DNA. Mutações no BRCA2 também estão ligadas a um alto risco de câncer de mama (tanto em homens quanto em mulheres, embora seja mais comum em mulheres) e câncer de ovário. Homens com mutações no BRCA2 têm um risco elevado de câncer de próstata e câncer de pâncreas também. Mulheres com uma mutação no BRCA2 têm até 70% de risco vitalício de câncer de mama e cerca de 20-30% de risco de câncer de ovário.
O gene BRIP1 (Proteína C-Terminal Helicase 1) está mapeado no cromossomo 17 (17q22) e codifica uma proteína que interage com BRCA1 e está envolvida no reparo de quebra de fita dupla de DNA de DNA. Mutações em BRIP1 estão associadas a um risco aumentado de câncer de ovário e potencialmente câncer de mama.
O gene CDH1 (Caderina 1) está mapeado no cromossomo 16 (16q22.1) e codifica uma proteína envolvida na adesão celular, sendo um supressor de tumor que previne a disseminação de células cancerosas. Mutações em CDH1 estão associadas ao câncer gástrico difuso hereditário e ao câncer de mama lobular. Faz parte da síndrome do câncer gástrico difuso hereditário (HDGC).
O gene CHEK2 (Checkpoint Kinase 2) está mapeado no cromossomo 22 (22q12.1) e codifica uma quinase que atua como checkpoint do ciclo celular, ajudando a reparar danos ao DNA e regulando o ciclo celular. Mutações em CHEK2 estão associadas a um aumento moderado nos riscos de câncer de mama, câncer colorretal e câncer de próstata.
O gene EPCAM (Epithelial Cell Adhesion Molecule) está mapeado no cromossomo 2 (2p21). E codifica uma proteína envolvida na adesão e migração celular, desempenhando um papel na regulação do reparo de incompatibilidade. Mutações no EPCAM podem levar à síndrome de Lynch, contribuindo para um risco aumentado de câncer colorretal e outros tipos de câncer, como câncer de endométrio e ovário.
O gene MLH1 (MutL Homólogo 1) está mapeado no cromossomo 3 (3p21) e codifica uma proteína que constitui um componente-chave do sistema de reparo de incompatibilidade, corrigindo erros de replicação de DNA. Mutações em MLH1 estão associadas à síndrome de Lynch, que aumenta o risco de câncer colorretal, câncer endometrial, câncer de ovário e outros tipos de câncer.
O gene MRE11 (MRE11A) está mapeado no cromossomo 11 (11q21) e codifica uma proteína envolvida no reparo de quebras de fita dupla de DNA e faz parte do complexo MRN (com RAD50 e NBS1). Mutações em MRE11 estão relacionadas ao câncer de mama e outros tipos de câncer devido a defeitos no reparo de DNA.
O gene MSH2 (MutS Homólogo 2) está mapeado no cromossomo 2 (2p21) e codifica proteína com papel central na via de reparo de incompatibilidade que detecta e repara erros na replicação do DNA. Mutações em MSH2 causam a síndrome de Lynch, aumentando significativamente o risco de câncer colorretal, endometrial, ovariano e outros.
O gene MSH6 (MutS Homólogo 6) está mapeado no cromossomo 2 (2p16) e codifica uma proteína que participa do sistema de reparo de incompatibilidade e trabalha com MSH2 para corrigir erros de replicação do DNA. Mutações em MSH6 também contribuem para a síndrome de Lynch, com riscos aumentados de câncer colorretal, câncer endometrial e outros tipos de câncer.
O gene NBN (Nibrin) está mapeado no cromossomo 8 (8q21) e codifica uma proteína que faz parte do complexo MRN e está envolvida na detecção e reparo de danos ao DNA. Mutações em NBN podem levar ao câncer de mama e aumentar o risco de leucemia e linfoma não-Hodgkin.
O gene NF1 (Neurofibromina 1) está mapeado no cromossomo 17 (17q11.2) e codifica uma proteína que ajuda a regular o crescimento celular ao inativar a via de sinalização Ras. Mutações em NF1 causam Neurofibromatose Tipo 1, que aumenta o risco de neurofibromas, tumores malignos da bainha do nervo periférico e outros tipos de câncer.
O gene PALB2 (Partner and Localizer of BRCA2) está mapeado no cromossomo 16 (16p12) e codifica uma proteína que trabalha com BRCA2 no reparo de DNA, estabilizando BRCA2 e promovendo recombinação homóloga. Mutações em PALB2 estão ligadas ao câncer de mama e câncer de ovário, com riscos ao longo da vida semelhantes aos das mutações BRCA2.
O gene PMS1 (Postmeiotic Segregation Increased 1) está mapeado no cromossomo 2 (2q31) e codifica uma proteína envolvida na via de reparo de incompatibilidade. Mutações em PMS1 podem levar à síndrome de Lynch, aumentando o risco de câncer colorretal e outros tipos de câncer.
O gene PMS2 (Postmeiotic Segregation Increased 2) está mapeado no cromossomo 7 (7p22) e codifica uma proteína que também desempenha um papel no reparo de incompatibilidade e trabalha com outras proteínas de reparo de incompatibilidade. Mutações em PMS2 levam à síndrome de Lynch, com um risco aumentado de câncer colorretal, câncer endometrial e outros.
O gene PTEN (Phosphatase and Tensin Homolog) está mapeado no cromossomo 10 (10q23.31) e codifica uma proteína que atua como supressora de tumor, regulando a divisão celular e impedindo que as células cresçam descontroladamente. Mutações PTEN estão associadas à síndrome de Cowden, aumentando o risco de câncer de mama, tireoide e endometrial.
O gene RAD50 está mapeado no cromossomo 5 (5q31) e codifica uma proteína que participa do complexo MRN envolvido no reparo de quebra de fita dupla de DNA. Mutações no RAD50 podem aumentar o risco de câncer de mama e outros tipos de câncer devido a mecanismos defeituosos de reparo de DNA.
O gene RAD51C está mapeado no cromossomo 17 (17q21) e codifica uma proteína que desempenha um papel no reparo de DNA auxiliando na recombinação homóloga. Mutações no RAD51C aumentam o risco de câncer de ovário e câncer de mama.
O gene RAD51D está mapeado no cromossomo 17 (17q12) e codifica uma proteína que está envolvida no reparo de danos ao DNA por meio de recombinação homóloga. Mutações no RAD51D estão associadas a um risco aumentado de câncer de ovário e câncer de mama.
O gene SMARCA4 (SWI/SNF Related, Matrix Associated Actin Dependent Regulator of Chromatin) está mapeado no cromossomo 19 (19p13.2) e codifica uma proteína que participa do complexo de remodelação da cromatina SWI/SNF, ajudando a controlar a expressão genética e o reparo do DNA. Mutações SMARCA4 estão ligadas ao carcinoma de pequenas células do ovário e potencialmente ao câncer de mama.
O gene STK11 (serina/treonina quinase 11) está mapeado no cromossomo 19 (19p13.3) e codifica uma quinase envolvida na regulação do crescimento e metabolismo celular. Mutações em STK11 causam a síndrome de Peutz-Jeghers, que aumenta o risco de câncer gastrointestinal, de mama, de ovário e de pâncreas.
O gene TP53 (Proteína Tumoral 53) está mapeado no cromossomo 17 (17p13) e codifica uma proteína que regula o ciclo celular e a apoptose, crucial para prevenir o desenvolvimento do câncer. Mutações em TP53 causam a síndrome de Li-Fraumeni, aumentando significativamente o risco de câncer de mama, sarcomas, tumores cerebrais, leucemia e outros tipos de câncer.
O gene XRCC2 (X-Ray Repair Cross Complementing 2) está mapeado no cromossomo 7 (7q11) e codifica uma proteína envolvida no reparo de danos ao DNA, especificamente no reparo de recombinação homóloga. Mutações em XRCC2 podem aumentar o risco de câncer de mama e outros tipos de câncer devido a defeitos nos mecanismos de reparo do DNA.
UTILIDADE DO EXAME:
Este teste deve ser solicitado pelo médico e visa:
• Identificar variantes alélicas da linha germinativa. Os tipos variantes detectados incluem variantes de base única (SNV), inserções / deleções pequenas (Indels) e variações no número de cópias (CNV)
• Avaliar do risco de câncer. Estimar o risco de câncer ao longo da vida para indivíduos com base na presença de variantes associadas a câncer hereditário.
• Gerenciamento personalizado do paciente. O sequenciamento de genes associados ao câncer hereditário pode ajudar a orientar o tratamento e o gerenciamento personalizados.
• Permitir o Aconselhamento genético e planejamento familiar. Os testes genéticos fornecem aos membros da família informações importantes sobre seus próprios riscos de câncer, permitindo intervenção precoce ou medidas preventivas.
• Detecção e triagem precoces. A identificação de mutações associadas a câncer hereditário pode levar a estratégias aprimoradas de triagem e detecção precoce.
• Participar de ensaios clínicos e pesquisa. Determinar a elegibilidade para ensaios clínicos adaptados a indivíduos com mutações genéticas específicas.
• Identificar as síndromes hereditárias do câncer. Ajuda a identificar indivíduos que herdaram mutações associadas a síndromes específicas.
• Avaliar o impacto de variantes de significância desconhecidas (VUS). Em alguns casos, análises adicionais e estudos familiares são necessários para determinar a significância clínica dessas variantes.
• Permitir a adoção de medidas de prevenção e profilaxia. Indivíduos com altos riscos genéticos podem escolher medidas preventivas, incluindo medicamentos (por exemplo, tamoxifeno para redução do risco de câncer de mama), profilaxia cirúrgica (por exemplo, mastectomia ou ooforectomia) ou mudanças no estilo de vida para reduzir o risco de câncer.
NECESSIDADE DE PEDIDO MÉDICO
O pedido médico deve ser enviado juntamente com a amostra.
COBERTO POR OPERADORA
Sim. A diretriz de utilização (DUT) nº 110 define os grupos para os quais a cobertura deste exame é obrigatória. Os subitens desta DUT contemplados por este exame são
110.07 – Câncer de Mama e Ovário Hereditários – Gene Brca1/Brca2
110.26 – Painel de Genes para Câncer de Mama e/ou Ovário
110.30 – Síndrome de Cowden
110.32 – Síndrome de Li-Fraumeni
110.33 – Síndrome de Lynch – Câncer Colorretal Não Poliposo Hereditário (HNPCC)
110.38 – Síndrome de Câncer Gástrico Difuso Hereditário
110.42 – Síndrome de Peutz-Jeghers
O código TUSS para este exame é 40503801 – Sequenciamento de Nova Geração (NGS) – genes isolados, painéis e grandes regiões genômicas (inclui Captura, Amplificação e Sequenciamento);
NECESSIDADE DE CONSENTIMENTO
Sim.
PREPARAÇÃO DA PACIENTE
Nenhuma preparação é necessária se a amostra for sangue. Para amostra de saliva ou swab 30 minutos antes da coleta, não beber, comer, fumar, mascar chicletes, escovar os dentes ou inserir qualquer objeto na boca.
O resultado é apresentado de forma simples e objetiva, a fim de facilitar sua compreensão. Contudo, a interpretação deste resultado é um ato médico e deve ser realizada levando-se em consideração os dados clínicos e demais exames do paciente.
Os resultados possíveis e suas interpretações estão apresentados a seguir.
DETECTADO. Foi detectada uma variante alélica em heterozigose denominada [nome da variante] no [nome do gene]. Este resultado indica que a paciente é portadora de uma variante patogênica ou provavelmente patogênica. Esta classificação pode ser obtida a partir do banco de dados ClinVar ou realizada com base em critérios definidos pela ACMG
NÃO DETECTADO. Não foram detectadas variantes alélicas patogênicas, provavelmente patogênicas no painel de genes testado. Este resultado indica que a paciente não é portadora de variantes classificadas atualmente como patogênicas ou provavelmente patogênicas neste painel gênico
DISPONIBILIDADE DO LAUDO (TAT)
O laudo estará disponível em até 30 dias corridos após a autorização do exame.
VALORES DE REFERÊNCIA
O valor de referência para este exame é NÃO DETECTADO.
PRECAUÇÕES
Variantes alélicas classificadas como Benignas/Possivelmente Benignas não serão reportadas, exceto sob solicitação expressa do médico assistente. A classificação e interpretação das alterações identificadas são realizadas com base no conhecimento atual e no consenso da comunidade científica no momento da elaboração do laudo. Resultados inconclusivos devem ser reavaliados periodicamente e podem ser reclassificados, de modo a incorporar as evidências científicas mais atuais. Classificações das variantes alélicas detectadas podem ser alteradas ao longo do tempo, à medida que novas informações científicas e clínicas se tornem disponíveis. Devido às limitações técnicas dos equipamentos e dos métodos de análise, podem ocorrer resultados falso-positivos ou falso-negativos.
NOTAS
Recomenda-se o aconselhamento genético antes e após o exame, com médico geneticista ou com profissional capacitado para avaliação genômica. Este laudo não deve ser copiado ou reproduzido, exceto em sua totalidade.
OBSERVAÇÕES
A Encondexa está comprometida em manter a confidencialidade das informações dos pacientes. Volume Mínimo (quantidade de espécime necessária para realizar um ensaio uma vez.
Recepção do volume mínimo torna impossível repetir o teste ou realizar testes de confirmação. Em algumas situações, um volume mínimo do espécime pode resultar em quantidade não suficiente, exigindo que um segundo espécime seja coletado).
Informe se o caso envolve “Resultados Semi-Urgentes” definidos como resultados relacionados a doenças infecciosas prontamente necessários para evitar consequências de saúde potencialmente sérias para o paciente.
Teste URGENTE: em raras circunstâncias, o teste URGENTE do laboratório de referência pode ser necessário para pacientes que precisam de tratamento imediato. Para agendar o teste URGENTE, peça ao patologista, médico ou gestor de laboratório ou Representante para ligar para a Encodexa™ e uma vez acordado que há necessidade da categoria de teste URGENTE, serão feitos arranjos para atribuir recursos para executar o teste em uma base URGENTE quando a amostra for recebida.
A Encodexa™ usa no mínimo dois identificadores específicos do paciente para verificar se o paciente correto é correspondido com o espécime correto e o pedido correto para serviços de teste. Conforme um espécime é recebido na ENCODEXA, o nome e sobrenome do paciente, data de nascimento, número do prontuário médico e número de acesso do cliente são verificados comparando os rótulos no tubo ou recipiente do espécime com o pedido eletrônico e qualquer papelada (folha de lote ou formulário) que possa acompanhar o espécime a ser testado. Quando discrepâncias são identificadas, o Centro de Atendimento de Consultas da Encondexa telefonará para o cliente para verificar as informações discrepantes para garantir que a Encodexa™ esteja realizando o teste correto para o paciente correto. Os espécimes são considerados rotulados incorretamente quando há uma incompatibilidade entre os identificadores específicos da pessoa no espécime e as informações que acompanham o espécime. Quando identificação insuficiente ou inconsistente for enviada, a Encodexa™ recomendará que um novo espécime seja obtido.
Prazo dos exames (TAT) O extenso menu de testes do Encodexa™ reflete as necessidades de nossa própria prática de assistência médica. Estamos comprometidos em fornecer o TAT mais rápido possível para melhorar o diagnóstico e o tratamento. Consideramos os serviços laboratoriais como parte do continuum de atendimento ao paciente, em que as necessidades do paciente são primordiais. Nesse contexto, nos esforçamos para cumprir nossas obrigações de serviço. Nosso histórico de serviço e nossas métricas de qualidade documentarão nossa capacidade de entregar em todas as áreas de serviço, incluindo TAT. A Encodexa™ define TAT como o tempo de teste analítico (o tempo do qual uma amostra é recebida no local de teste até o momento do resultado) necessário e é listado para cada teste como “Relatório disponível”. O TAT é monitorado continuamente por cada local de laboratório em execução dentro da Encodexa™.
TIPO
Amostra de sangue periférico, EDTA tubo com tampa violeta. Não é necessário jejum.
Para outras amostras, contatar a Genoa/LPCM.
INSTRUÇÕES DE COLETA E ARMAZENAMENTO DA AMOSTRA
O sangue deve ser colhido em tubo a vácuo contendo EDTA (tampa violeta)
ESTABILIDADE DA AMOSTRA
Armazenar sob refrigeração (2-8ºC). Nestas condições, a amostra é estável por 5 dias.
VOLUME MÍNIMO DA AMOSTRA
Volume ideal (3ml); volume mínimo (1ml).
RETENÇÃO DA AMOSTRA
O sangue é descartado após 7 dias; o DNA extraído é descartado após 180 dias.
INSTRUÇÕES DE COLETA DE AMOSTRAS
1. A amostra standard recomendada é de 4 ml de sangue total obtido por punção venosa em vacutainer K2EDTA tampa violeta;
2. Para fase pré-natal quando por exemplo exames de ultrassonografia do feto ou testes bioquímicos apresentarem alterações a amostra preferencial é líquido amniótico ou biópsia de vilosidade corial;
3. Para FIV (Fecundação in vitro) são retiradas células do ovo que será implantado no endométrio PGT-A (Teste Genético Pré-Implantacional para Aneuploidias). O material pode ser submetido a diversas metodologias como NGS ou array CGH SNP;
Para avaliação do DNA de embrião existem três abordagens para obtenção de amostras:
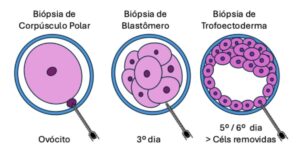
Estas abordagens como mostra o diagrama acima incluem biópsia do primeiro ou segundo corpúsculos polares do ovócito, biópsia dos blastômeros no terceiro dia de desenvolvimento do embrião e finalmente a mais aceita atualmente a biópsia do trofoectoderma (TE) do blastocisto no quinto ou sexto dia de desenvolvimento. Nesta última, mais células podem ser removidas nesse estágio e potencialmente permite diagnóstico mais preciso.
NECESSIDADE DE PEDIDO MÉDICO
Sim, informações clínicas presentes na requisição são extremamente importantes para corroborar na análise do array CGH SNP e para enriquecimento do banco de dados.
NECESSIDADE DE CONSENTIMENTO
Para array CGH SNP há necessidade de Consentimento do paciente ou pessoa responsável pelo paciente se este apresentar alguma incapacidade intelectual. Estes exames podem revelar inconsistências parenterais.
